Présentation
RÉSUMÉ
Cet article traite de la modélisation numérique du comportement de matériaux cristallins. Après une description des modèles de simulations à l’échelle atomique et des applications à l’étude de la déformation plastique des métaux, l’article aborde les modèles numériques du comportement collectif de populations de dislocations pour lesquels les entités simulées sont les lignes de défauts. Puis, deux modèles d'étude du comportement du monocristal sont présentés. Le premier utilise les densités de dislocations sur les systèmes de glissement, tandis que le deuxième est purement phénoménologique et permet de traiter des chargements complexes.
Lire cet article issu d'une ressource documentaire complète, actualisée et validée par des comités scientifiques.
Lire l’articleAuteur(s)
-
Marc FIVEL : Agrégé de mécanique de l’École normale supérieure de Cachan - Docteur en mécanique - Chargé de recherches au CNRS - Institut national polytechnique de Grenoble
-
Samuel FOREST : Ingénieur civil de l’École des mines de Paris - Docteur en sciences et génie des matériaux - Chargé de recherches au CNRS - École nationale supérieure des mines de Paris
INTRODUCTION
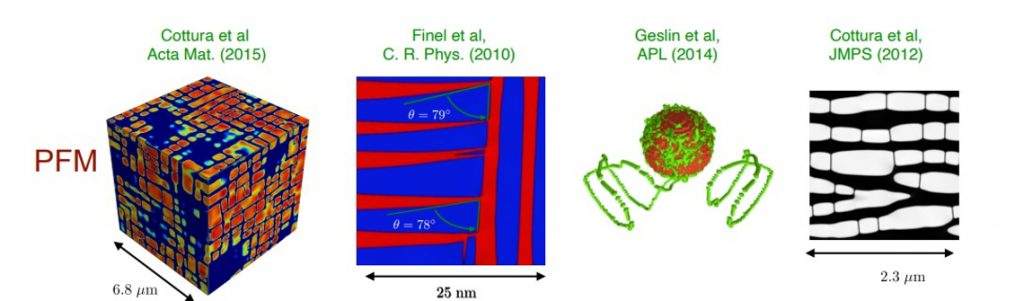
Les matériaux cristallins sont par nature hétérogènes. On distingue, par exemple, les polycristaux constitués d’agglomérats de grains monocristallins dans lesquels des lignes de défauts, les dislocations, propagent des cisaillements sur des plans cristallographiques. Cette hétérogénéité spatiale conduit à développer des modélisations différentes à chacune des échelles impliquées. L’avénement d’une puissance informatique toujours plus performante (on peut actuellement compter sur une vitesse des microprocesseurs doublée tous les dix-huit mois) a stimulé le développement d’outils numériques sophistiqués dédiés à la simulation du comportement mécanique des matériaux. À chaque échelle d’étude, un effort important a été porté sur la mise en place de modèles fiables adaptés aux diverses situations rencontrées lors des procédés de mise en forme et des conditions en service des composants industriels (figure 1). Petit à petit, chaque modèle devient de plus en plus performant : le volume qu’il est possible de simuler ainsi que le temps physique sont toujours plus importants. On constate désormais un recouvrement entre les capacités des différents modèles à simuler la réponse de volumes de taille fixée pendant un temps physique donné.
Même s’il est encore utopique de penser simuler le processus de mise en forme par emboutissage à partir de simulations atomiques ou même à partir de la dynamique des dislocations, on constate tout de même que l’on peut, d’ores et déjà, « remonter » les échelles d’espace et de temps de manière continue, par exemple en réalisant des simulations spécifiques dont les résultats serviront à asseoir un modèle à l’échelle supérieure. On observe également que, à l’échelle des milieux continus, les lois de comportement phénoménologiques laissent peu à peu la place à des relations de comportement déduites des mécanismes physiques à l’origine de la déformation plastique tels que les mouvements de dislocations. Cette transition d’échelle entre la dynamique d’une ligne de dislocation et le comportement d’un milieu continu pour lequel les variables internes sont généralement les densités de dislocations sur les différents systèmes de glissement implique une moyenne et donc une statistique sur tous les événements potentiels ainsi qu’une homogénéisation sur un volume arbitraire. Cela conduit finalement à des relations de comportement toujours plus ou moins phénoménologiques. Il en est de même pour les techniques d’homogénéisation modélisant le passage du monocristal au polycristal.
Le présent article est consacré à la modélisation numérique du comportement de matériaux cristallins, principalement métalliques, à différentes échelles à l’aide d’outils numériques spécifiquement adaptés en insistant sur les applications potentielles de ces outils, leurs capacités à reproduire la déformation plastique des cristaux mais également leurs limitations intrinsèques. Après une description des modèles de simulations à l’échelle atomique, comme la dynamique moléculaire, et des applications concernant l’étude de la déformation plastique des métaux, l’article aborde les modèles numériques du comportement collectif de populations de dislocations pour lesquels les entités simulées sont les lignes de défauts. Puis, le comportement du monocristal est décrit en présentant deux modèles : le premier utilise comme variables internes les densités de dislocations sur les systèmes de glissement tandis que le deuxième est purement phénoménologique mais remarquablement bien adapté pour traiter des chargements complexes.
Dans un second article , nous traiterons le cas du polycristal.
Cet article est réservé aux abonnés.
Il vous reste 92 % à découvrir.
Déjà abonné ? Se connecter
DOI (Digital Object Identifier)
Présentation
Article inclus dans l'offre
"Étude et propriétés des métaux"
(200 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.
1. Simulations à l’échelle atomique
Ce paragraphe décrit les notions fondamentales des modèles de dynamique moléculaire utilisés pour traiter la plasticité des métaux. Le lecteur désireux d’en connaître davantage sur la méthode peut consulter la référence [1] parue dans les Techniques de l’Ingénieur.
1.1 Potentiels interatomiques
Les propriétés physiques et mécaniques des matériaux découlent directement de leur structure atomique et de la nature des liaisons entre atomes. On distingue cinq familles de liaisons interatomiques :
-
la liaison ionique que l’on retrouve par exemple dans les céramiques (MgO, Al2O3) ou dans les cristaux ionique (NaCl, CsCl) ;
-
la liaison covalente qui se rencontre dans les polymères et le diamant (liaison carbone‐carbone) et également dans les semi‐ conducteurs utilisés en microélectronique Si, Ge, GaAs ;
-
la liaison métallique caractéristique des métaux (Cu, Al, Ag, Na, Fe, Mo, Ta, Ti…).
À ces trois liaisons fortes s’ajoutent deux liaisons plus faibles :
-
la liaison de Van der Walls à l’origine de la cohésion des cristaux de gaz rares (Ar, Ne, Xe, Kr) ;
-
la liaison hydrogène responsable de la cohésion de la glace et que l’on rencontre également entre chaînes de polymères.
Modéliser la déformation plastique des cristaux à l’échelle de l’atome nécessite de modéliser les énergies d’interaction agissant entre les différents constituants du matériau sachant qu’il s’agit presque toujours d’une combinaison des différentes liaisons. Ces énergies, appelées potentiels interatomiques, sont au cœur des modèles numériques de dynamique moléculaire présentés au paragraphe 1.2. De façon générale, les potentiels interatomiques comportent un terme de répulsion à courte distance complété par un terme d’attraction à plus grande distance. La répulsion entre atomes provient du principe quantique d’exclusion de Pauli et se modélise de façon phénoménologique par une fonction rapidement décroissante, typiquement en 1/r...
Cet article est réservé aux abonnés.
Il vous reste 93 % à découvrir.
Déjà abonné ? Se connecter
Simulations à l’échelle atomique
Article inclus dans l'offre
"Étude et propriétés des métaux"
(200 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.
BIBLIOGRAPHIE
-
(1) - CHANTRENNE (P.), VOLZ (S.) - Thermique à l’échelle submicronique. Introduction à la dynamique moléculaire. - Techniques de l’Ingénieur, traité Génie énergétique, BE 8 290, p. 1-20 (2002).
-
(2) - DAW (M.S.), FOILES (S.M.), BASKES (M.I.) - The embedded-atom method : a review of theory and applications. - Materials Science Report, 9, p. 251-310 (1993).
-
(3) - ABRAHAM (F.) - The embedded-atom method : a review of theory and applications. - Materials Science Report, 9, p. 251-310 (2002).
-
(4) - TADMOR (E.B.), ORTIZ (M.), PHILLIPS (R.) - Quasicontinuum analysis of defects in solids. - Philosophical Magazine, A73, p. 1529-1563 (1996).
-
(5) - SHENOY (V.B.), MILLER (R.), TADMOR (E.B.), RODNEY (D.), PHILLIPS (R.), ORTIZ (M.) - An adaptative finite element approach to atomistic-scale mechanics - the quasicontinuum method. - Journal of the Mechanics and Physics of Solids, 47, p. 611-642 (1999).
-
...
Cet article est réservé aux abonnés.
Il vous reste 93 % à découvrir.
Déjà abonné ? Se connecter
Article inclus dans l'offre
"Étude et propriétés des métaux"
(200 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.