


Présentation
Auteur(s)
-
Yves JEANNIN : Ingénieur de l’École Nationale Supérieure de Chimie de Paris - Professeur à l’Université Pierre et Marie Curie - Correspondant de l’Institut
Lire cet article issu d'une ressource documentaire complète, actualisée et validée par des comités scientifiques.
Lire l’articleINTRODUCTION
Avant de décrire les différentes phases de la résolution d’une structure, il est bon de rappeler que le modèle théorique utilisé pour établir les méthodes de résolution implique des hypothèses qu’il faut formuler précisément afin de bien voir les limites de la méthode
Les atomes ont une distribution électronique sphérique. Cette hypothèse se retrouve dans les tables donnant le facteur atomique de diffusion. On sait que les rayons X interagissent avec les électrons : en quelque sorte, ils comptent les électrons des différents atomes et l’on obtient le barycentre du nuage électronique de chaque atome. Ce point n’a pas de raison de coïncider avec le noyau ; toutefois, l’écart est d’autant plus petit que l’atome a un numéro atomique plus élevé. Dans ces conditions, les distances mesurées entre atomes légers, Li et H par exemple, même si la méthode les donne avec une précision de 0,001Å (1Å = 0,1 nm), ne sauraient être considérées sans un certain discernement.
Notons que les neutrons ignorent cette difficulté car ils interagissent avec les noyaux.
Les vibrations thermiques des atomes sont considérées comme centrosymétriques mais pas forcément isotropes. En fait, le temps d’observation est très long par rapport à la période de vibration si bien que la densité électronique observée est une moyenne dans le temps. Cette remarque a une conséquence : il sera impossible de distinguer par rayons X le désordre statique du désordre dynamique ; le désordre statique est lié, par exemple, à l’occupation d’un même site cristallographique par deux atomes ayant des dimensions analogues mais des numéros atomiques différents ; le désordre dynamique est celui d’un groupe d’atomes en mouvement rapide dans le cristal, par exemple un groupe CH3 tournant autour de son axe ternaire.
Les atomes sont considérés comme vibrant indépendamment les uns des autres. La spectrométrie infrarouge nous apprend que cela n’est pas vrai. Des tentatives, au prix d’un effort mathématique important, ont été faites pour cerner la réalité de plus près. Il ne semble pas que le prix à payer en vaille la peine. Parfois, lorsque l’on étudie une molécule dont l’organisation des liaisons lui confère un caractère particulièrement rigide, comme le camphre, on peut mettre en œuvre un modèle où la molécule est prise comme un seul bloc rigide.
Enfin, le modèle suppose un cristal parfait infini. Le cristal examiné est forcément fini, tout au plus 2 mm dans sa plus grande dimension. Par ailleurs, le cristal possède en fait une structure dite mosaïque, il est dit idéalement imparfait ; il est fait de parcelles dont la dimension est de l’ordre du micromètre et qui sont désorientées par rapport à leurs voisines de quelques secondes d’arc. L’orientation moyenne de ces blocs correspond à l’orientation du cristal. Chaque bloc est parfait et diffracte indépendamment de ses voisins. Cette situation particulière permet l’application de la théorie cinématique qui établit que l’intensité diffractée par un plan est proportionnelle au carré du module du facteur de structure.
Le lecteur se reportera utilement à l’article « Cristallographie géométrique » du traité Sciences fondamentales.
Pour une utilisation plus pratique, le lecteur pourra se reporter utilement à l’article « Détermination de structure cristalline par rayons X : méthodes numériques » [24].
Cet article est réservé aux abonnés.
Il vous reste 93 % à découvrir.
Déjà abonné ? Se connecter
VERSIONS
- Version archivée 1 de juil. 1983 par Yves JEANNIN
DOI (Digital Object Identifier)
Présentation
Article inclus dans l'offre
"Techniques d'analyse"
(288 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.
5. Dépouillement de la structure
5.1 Repérage des atomes lourds
Même si les positions de tous les atomes ne sont pas déduites de la fonction de Patterson, le repérage des atomes lourds permet de résoudre la structure.
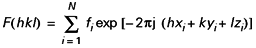
fi est fonction du nombre d’électrons de l’atome i alors que l’exponentielle conduit à un cosinus et un sinus toujours inférieurs à 1. Le f de l’atome lourd est important puisqu’il dépend du nombre d’électrons. Il y a donc de grandes chances pour que la phase calculée à partir du (ou des) seul atome lourd soit suffisamment approchée.
Si donc, on calcule une série de Fourier à l’aide des F mesurés auxquels on affecte la phase déduite d’un calcul de F partiel basé sur l’atome lourd, on verra apparaître, outre les atomes connus, les autres atomes de la structure. Les hauteurs des pics ne correspondront peut-être pas exactement aux nombres d’électrons attendus, mais les distances et les angles interatomiques permettront de reconnaître la structure sur laquelle on possède toujours chimiquement des éléments d’information. Si tous les atomes ne sont pas vus, le procédé sera répété : affinement des positions des atomes repérés, calcul des F sur cette base, affectation des phases trouvées aux F mesurés, nouveau calcul d’une série de Fourier et ainsi de suite.
HAUT DE PAGE5.2 Facteur de reliabilité
On peut suivre l’avancement de la résolution et surtout sa validité, à l’aide du facteur R dit de reliabilité :
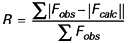
Si les observations sont pondérées à l’aide d’un facteur w, on calcule un facteur R pondéré :
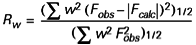
Cet article est réservé aux abonnés.
Il vous reste 93 % à découvrir.
Déjà abonné ? Se connecter
Dépouillement de la structure
Article inclus dans l'offre
"Techniques d'analyse"
(288 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.
Bruker
Nonius France
Huber Diffraktionstechnik
Osmic
Oxford Instruments
Stoe
X-Ray Research
HAUT DE PAGE
###
Ouvrages de références
International tables for X-Ray cristallography - . Vol. I (1969), Vol. II (1972), Vol. III (1968), Vol. IV (1974). Kynoch Press.
Ouvrages généraux
BUERGER (M.) - X-ray crystallography - . 1962 Wiley.
WOOLFSON (M.M.) - X-ray crystallography - . 1970 Cambridge University Press.
LADD (M.F.C.) - PALMER (R.A.) - Structure determination by X-ray crystallography - . 1977 Plenum Press.
BUERGER (M.) - Crystal structure analysis - . 1967 Wiley.
BUERGER (M.) - Vector space - . 1959 Wiley.
GUINIER (A.) - Théorie et pratique de la radiocristallographie - . 1956 Dunod.
JAMES (R.W.) - The optical principles of the diffraction of X-rays - . 1954 Bell and Sons.
LIPSON (H.) - COCHRAN (W.) - The determination of crystal structures - . 1954 Bell and Sons.
Méthodes directes
LADO (M.F.C.) - PALMER (R.A.) - Theory and practice of direct methods in crystallography - . 1980 Plenum Press.
AHMED (F.R.) - Crystallographic computing techniques - . 1976 Munksgaard.
Calculs cristallographiques
ROLLETT (J.S.) - Computing methods in crystallography - . 1965...
Cet article est réservé aux abonnés.
Il vous reste 94 % à découvrir.
Déjà abonné ? Se connecter
Article inclus dans l'offre
"Techniques d'analyse"
(288 articles)
Actualisée et enrichie d’articles validés par nos comités scientifiques.
Quiz, médias, tableaux, formules, vidéos, etc.
Opérationnels et didactiques, pour garantir l'acquisition des compétences transverses.
Un ensemble de services exclusifs en complément des ressources.






